
Stel: bij een uitbraak van salmonella-besmettingen vindt en isoleert men de boosdoener, de specifieke stam. Dan nog valt er een hele weg af te leggen om die volledig tot zijn oorspronkelijke bron terug te traceren en op basis daarvan gepaste maatregelen te treffen. Via ‘whole genome sequencing’ van de stam en een ‘machine learning’ algoritme kan dat zoekproces voortaan een stuk sneller.
Salmonella-besmettingen: de dader laat zich niet makkelijk identificeren
Jaarlijks worden in België zowat 4.000 gevallen van salmonella gerapporteerd waarbij de bacterie via opname door voedsel een ontsteking van het darmslijmvlies veroorzaakt (FAVV website voor de consumenten). Vaak betreft dit individuele gevallen waarvoor men zich kan behoeden door een goede hygiëne toe te passen (het wassen van handen, messen en werkoppervlakken). Diverse types voedsel kunnen een besmettingsbron vormen, waaronder rauw vlees. Salmonella-ziekte is dan ook gekend als een zoönose (een infectie die van nature overdraagbaar is van gewervelde dieren - vaak symptoomloze dragers - op mensen). Vandaar dat het aangeraden is om vleesstukken steeds goed te doorbakken.
Af en toe durven er zich wel eens uitbraken van salmonella-besmettingen voordoen. Dit zijn gevallen van collectieve voedseltoxi-infectie (CVTI), waarbij onder dezelfde omstandigheden meerdere personen gelijkaardige symptomen vertonen en er een (waarschijnlijk) oorzakelijk verband bestaat met eenzelfde voedselbron. Om deze te herkennen (door de verschillende incidenten aan elkaar te linken) en hun oorzaak (oorspronkelijke besmettingsbron) te achterhalen zijn onderzoeken naar bacteriën op stamniveau nodig. Klassieke bacteriologie wordt daarbij gecombineerd met serologische testen. Voor salmonella bestaan er echter zo'n 2.600 verschillende serotypen. Kortom, dergelijke onderzoeken nemen heel wat tijd in beslag. Om het aantal serologische testen te beperken wordt vandaag de dag reeds beroep gedaan op snelle DNA fingerprintmethoden zoals Rep-PCR (gericht naar repetitieve DNA elementen die variëren in lengte) en pulsed field-gel electroforese (pulsotypes). De ultieme manier van identificatie bestaat er in om de DNA sequentie van het hele genoom te gaan bepalen. Met de snelle evoluties in ‘whole genome sequencing’ (WGS) is dit niet langer ondenkbaar.
Genoomverwantschappen binnen salmonella typhimurium
WGS wordt ondertussen volop toegepast op Salmonella. Zo zijn er voor de serovar Typhimurium reeds volledige genomen gekend van meer dan duizend isolaten, die afkomstig zijn van diverse bronnen: pluimvee, varkens, runderen, wilde vogels, mensen, levensmiddelen, etc. Recent brachten Amerikaanse onderzoekers (Zhang et al, 2019) de verwantschap (obv de DNA sequentie similariteit) van al die genomen in kaart en onderzochten of bepaalde groepen van verwante genomen in relatie konden gebracht worden met specifieke bronnen. Dat bleek het geval te zijn voor Typhimurium varianten die geïsoleerd waren uit pluimvee, varkens, runderen en wilde vogels (Figuur 1). Waarschijnlijk is dit het gevolg van adaptatie van die Typhimurium varianten aan een specifieke gastheer (of waard).
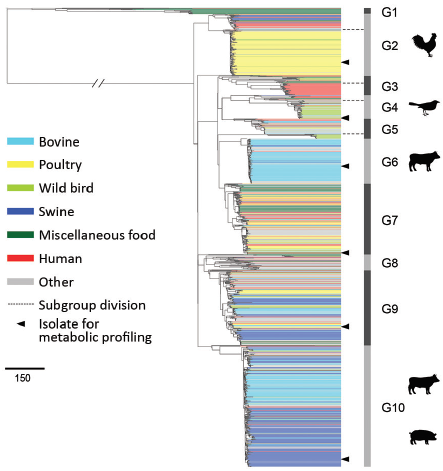
Figuur 1. Fylogenetische verwantschapsanalyse deelt de genomen van 1.267 Salmonella Typhimurium isolaten op in 10 groepen. Bepaalde subgroepen zijn duidelijk geassocieerd met een specifieke bron: G2b met pluimvee, G4b en G5b met wilde vogels, G6 met rund en G10 met zowel varken als rund. [bron: Zhang et al, 2019]
Een eerste manier om de mogelijke bron van een Salmonella Typhimurium isolaat te voorspellen is door zijn genoomsequentie mee op te nemen in de fylogenetische verwantschapsanalyse en te kijken in welke groep het zich plaatst. Als test pasten Zhang et al (2019) dit toe op genomen van isolaten van 8 belangrijke zoönotische salmonella-uitbraken die zich in de VS tussen 1998 en 2013 hebben voorgedaan. In 6 gevallen werd de bron ervan correct voorspeld.
Automatisch leeralgoritme voorspelt meest plausibele bron voor zoönotische salmonella
De onderzoekers ontwikkelden vervolgens via een ‘machine learning’ benadering een algoritme dat dierlijke bronnen kan voorspellen op basis van 3.137 genoomeigenschappen (puntmutaties, inserties/deleties en geselecteerde genen). De ‘Random Forest classifier’ werd getraind met genomen van isolaten afkomstig van pluimvee, varkens, runderen en wilde vogels. De classifier presteerde het best in het voorspellen van pluimvee en varken als bron (Figuur 2). In een test was het in staat om voor 7 van de 8 zoönotische salmonella-uitbraken de bron (gastheer) correct toe te wijzen.
De classifier werd ook toegepast op de genomen van humane Typhimurium isolaten. Zowat 32% van deze isolaten waren volgens de classifier zoönotisch en werden stuk voor stuk toegewezen aan ofwel pluimvee, varkens, runderen of wilde vogels als oorspronkelijke gastheer. Uitbreiding van de dataset met genomen van Typhimurium isolaten uit diverse bronnen (waaronder zeevruchten) is nodig om meer humane Typhimurium isolaten aan een bron toe te kunnen wijzen. Een moeilijkheid wordt evenwel gevormd door bepaalde Salmonella Typhimurium stammen die in staat zijn om in verschillende types gastheren te circuleren.

Figuur 2. Waarschijnlijkheid waarmee Typhimurium isolaten door de ‘Random Forest classifier’ correct kunnen worden toegewezen aan varken (blauw), pluimvee (geel), rund (cyaan) en wilde vogels (groen) als bron. [bron: Zhang et al, 2019]
Het automatisch leeralgoritme bracht ook specifieke genen naar boven die een grote bijdrage leverden bij het toewijzen van een isolaat aan een bron. Het betrof Salmonella genen die een rol spelen in interacties met de gastheer (virulentiegenen en genen betrokken bij maag-darm kolonisatie), in resistentie tegen koper (mineraal veevoederadditief) en in de ontwikkeling van de zweepstaart (fliC gen). Focus op deze genen samen met uitbreiding van de database, zou het toewijzingsmodel in de toekomst nog efficiënter kunnen maken.
BRONNEN
Zhang S. et al. (2019) Zoonotic Source Attribution of Salmonella enterica Serotype Typhimurium Using Genomic Surveillance Data, United States. Emerging Infectious Diseaeses 25, 82-91


